Chapter 2 Rarefaction Practice in Microbiome Data Analysis
Presented at the DataScience Group meeting, Quadram Institute Bioscience, July 2023
Co-author of this chapter: Andrea Telatin2
In the last decade, there has been ongoing debate regarding the optimal approach for normalizing sequencing depth in samples analyzed using 16S rRNA gene sequencing. A highly influential paper by McMurdie and Holmes (McMurdie and Holmes, 2014) strongly argued against the practice of rarefying sequence counts, deeming it “inadmissible” and discouraging its utilization. Despite a rebuttal on this subject (Weiss et al., 2017), which demonstrated the benefits of rarefying in certain cases, the proponents of alternative normalization method seem to have a predominant influence within the microbiome research community. Nevertheless, a recent publication by Patrick Schloss (Schloss, 2023), shows that rarefaction is the most robust approach to control for uneven sequencing effort when considered across a variety of alpha and beta diversity metrics.
2.1 Aims of this chapter
Collect some definitions and clarify the difference between the terms “rarefy” and “rarefaction”;
exemplify the previous terms in alpha and beta diversity analyses;
provide the code to implement rarefaction curves and to estimate rarefaction estimates of richness and Bray-Curtis dissimilarities.
2.2 Required packages
List of libraries used:
- mia (
BiocManager::install("mia")) - miaViz (
BiocManager::install("miaViz")) - ape (
install.packages("ape")) - scater (
BiocManager::install("scater")) - patchwork (
install.packages('patchwork')) - ranacapa (
devtools::install_github("gauravsk/ranacapa")) - BiocParallel (
BiocManager::install("BiocParallel")) - TreeSummarizedExperiment (
BiocManager::install("TreeSummarizedExperiment"))
To install ranacapa you will need devtools (package) and phyloseq from bioconductor.
2.3 Introduction and definitions
Rarefaction in microbiome research was introduced from ecology research out of the need to address the issue of uneven sampling depth. When studying microbial communities using high-throughput sequencing techniques like 16S rRNA gene sequencing or shotgun metagenomics, the number of sequencing reads obtained from different samples can vary significantly. This uneven sampling depth can introduce biases and make it challenging to compare the diversity and abundance of microbial taxa between samples accurately.
Rarefaction standardises the sampling effort across all samples and should ensure fair comparisons of diversity and abundance. By repeatedly subsampling the sequencing data to a common sequencing depth for each sample, the rarefaction method allows researchers to obtain a consistent and unbiased estimate of the microbial diversity present in each sample.
Library size normalization by random subsampling without replacement is called rarefying. There is confusion in the literature regarding terminology, and sometimes this normalization approach is conflated with a non-parametric resampling technique, called rarefaction. According to (McMurdie and Holmes, 2014), rarefying is most often defined by the following steps:
Select a minimum library size, \(N_{L,min}\). This has also been called the rarefaction level.
Discard libraries (microbiome samples) that have fewer reads than \(N_{L,min}\).
Subsample the remaining libraries without replacement such that they all have size \(N_{L,min}\).
Let’s make a practical example in R. We start by creating a very simple count matrix with 3 taxa and 2 samples.
# Generate a very simple count matrix where the first
# sample has half the library size of the second sample
count_matrix <- matrix(c(68, 32, 200, 200, 200, 200),
nrow = 3, ncol = 2)
colnames(count_matrix) <- c("S1", "S2")
rownames(count_matrix) <- c("Taxa1", "Taxa2", "Taxa3")
count_matrix## S1 S2
## Taxa1 68 200
## Taxa2 32 200
## Taxa3 200 200S2 sample has a library size which is twice the library size of sample S1.
## S1 S2
## 300 600According to the definition, we choose \(N_{L,min} = 300\) and we subsample, without replacement (random number generation seed equal to 123, see why), the counts such that they all the samples have size \(300\).
# Function to rarefy counts of a count matrix
rarefy_function <- function(count_matrix) {
# Determine the minimum library size across samples
minLibSize <- min(colSums(count_matrix))
# Perform rarefaction on each sample in the count matrix
count_rarefy <- apply(count_matrix, 2, FUN = function(smpl){
# Create a vector of taxon names proportional to their counts in the sample
vec <- rep(rownames(count_matrix), times = smpl)
# Perform subsampling by randomly selecting taxon names without replacement
subsampling <- sample(vec, size = minLibSize, replace = FALSE)
# Create a table of the subsampled taxon counts
table(subsampling)
})
return(count_rarefy)
}
# Set the random seed for reproducibility
set.seed(123)
rarefied_counts <- rarefy_function(count_matrix)
rarefied_counts## S1 S2
## Taxa1 68 103
## Taxa2 32 109
## Taxa3 200 88The resulting table is a table where the S1 remains the same as before subsampling. Indeed, subsampling \(N_{L,min} = 300\) counts from a sample with that same library size is like taking the entire sample. S2 instead, has now a total of 300 counts divided across the three taxa.
As the subsampling step is performed only once, another way to obtain the rarefied counts more easily, is to divide the counts by their library size, multiply the relative abundances by \(N_{L,min} = 300\), and round the results.
rarefy_simple_function <- function(count_matrix) {
# Determine the minimum library size across samples
minLibSize <- min(colSums(count_matrix))
# Perform simple rarefaction on each sample in the count matrix
count_rarefy_simple <- apply(count_matrix, 2, FUN = function(smpl){
# Calculate the relative abundance of each taxon in the sample
rel_abundance <- smpl / sum(smpl)
# Multiply the relative abundances by Lmin to obtain the rarefied counts
rarefied_counts <- round(minLibSize * rel_abundance, digits = 0)
# Return the rarefied counts for the current sample
return(rarefied_counts)
})
return(count_rarefy_simple)
}
count_rarefied_simple <- rarefy_simple_function(count_matrix)
count_rarefied_simple## S1 S2
## Taxa1 68 100
## Taxa2 32 100
## Taxa3 200 100According to (Schloss, 2023), the authors of (McMurdie and Holmes, 2014) were correct to state that the distinction between “rarefying” and “rarefaction” was confusing and led to their conflation. However, they poorly managed to solve the problem due to misleading sentences throughout the publication. Traditionally, repeating the subsampling step a large number of times and averaging the result is called rarefaction. Instead, rarefying or subsampling is rarefaction, but with a single randomization. To minimize confusion, we will use “subsampling” in place of “rarefying” through the rest of this chapter. For clarity, we will use the same definition of rarefaction of (Schloss, 2023):
Select a minimum library size, \(N_{L,min}\). Researchers are encouraged to report the value of \(N_{L,min}\).
Discard samples that have fewer reads than \(N_{L,min}\).
Subsample the remaining libraries without replacement such that they all have size \(N_{L,min}\).
Compute the desired metric (e.g., richness, Shannon diversity, Bray-Curtis distances) using the subsampled data.
Repeat steps 3 and 4 a large number of iterations (typically 100 or 1,000). Researchers are encouraged to report the number of iterations.
Compute summary statistics (e.g., the mean) using the values from step 4.
2.4 Rarefaction in practice
The rarefaction, i.e. repeating the subsampling step a large number of times and averaging the result, is here exemplified for Richness index and Bray-Curtis dissimilarity index.
Let’s start by using an example dataset from a research published in PNAS in early 2011 (Caporaso et al., 2011). This work compared the microbial communities from 25 environmental samples and three known “mock communities” – a total of 9 sample types – at a depth averaging 3.1 million reads per sample. The dataset comes with the mia package.
library(mia)
data("GlobalPatterns", package = "mia")
# See https://microbiome.github.io/mia/reference/agglomerate-methods.html
gp_genus <- mia::agglomerateByRank(GlobalPatterns, "Genus")
# Filter out rows (taxa) with zero counts across all samples
gp_genus <- gp_genus[
rowSums(assay(gp_genus, "counts")) > 0, ]
library(scater)
gp_genus <- addPerCellQC(gp_genus)Here we show the distribution of library sizes across samples. We have library sizes ranging from 58688 reads to 40x times more.
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 58688 567103 1106849 1085257 1527330 2357181# Create a scatter plot of the 'gp_genus' dataset, where the y-axis represents the 'sum' column in the colData,
# the x-axis represents the 'SampleType' column in the colData, and the points are colored by the 'SampleType' column
plotColData(object = gp_genus,
y = "sum", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1))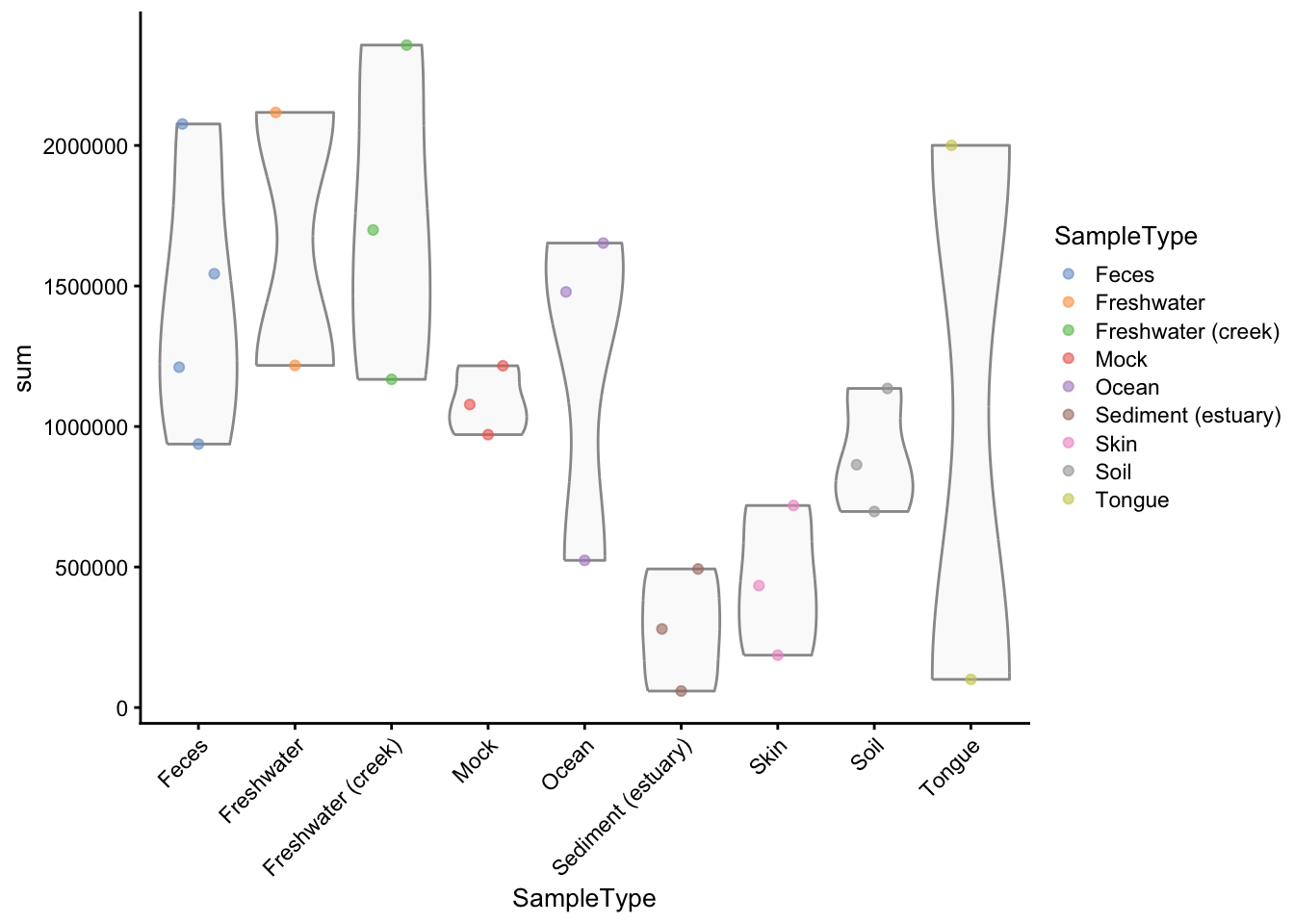
Figure 2.1: Sample library sizes grouped and coloured by sample type.
2.4.1 Richness index
Richness refers to the overall count of species within a community (sample). The most basic richness index corresponds to the number of observed species (observed richness). Richness estimates remain unaffected by the abundances of individual species.
To compute richness we use the estimateRichness function of the mia package.
We can inspect the observed richness distribution of each sample type:
p_r <- plotColData(object = gp_genus,
y = "observed", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Richness in raw counts")
p_r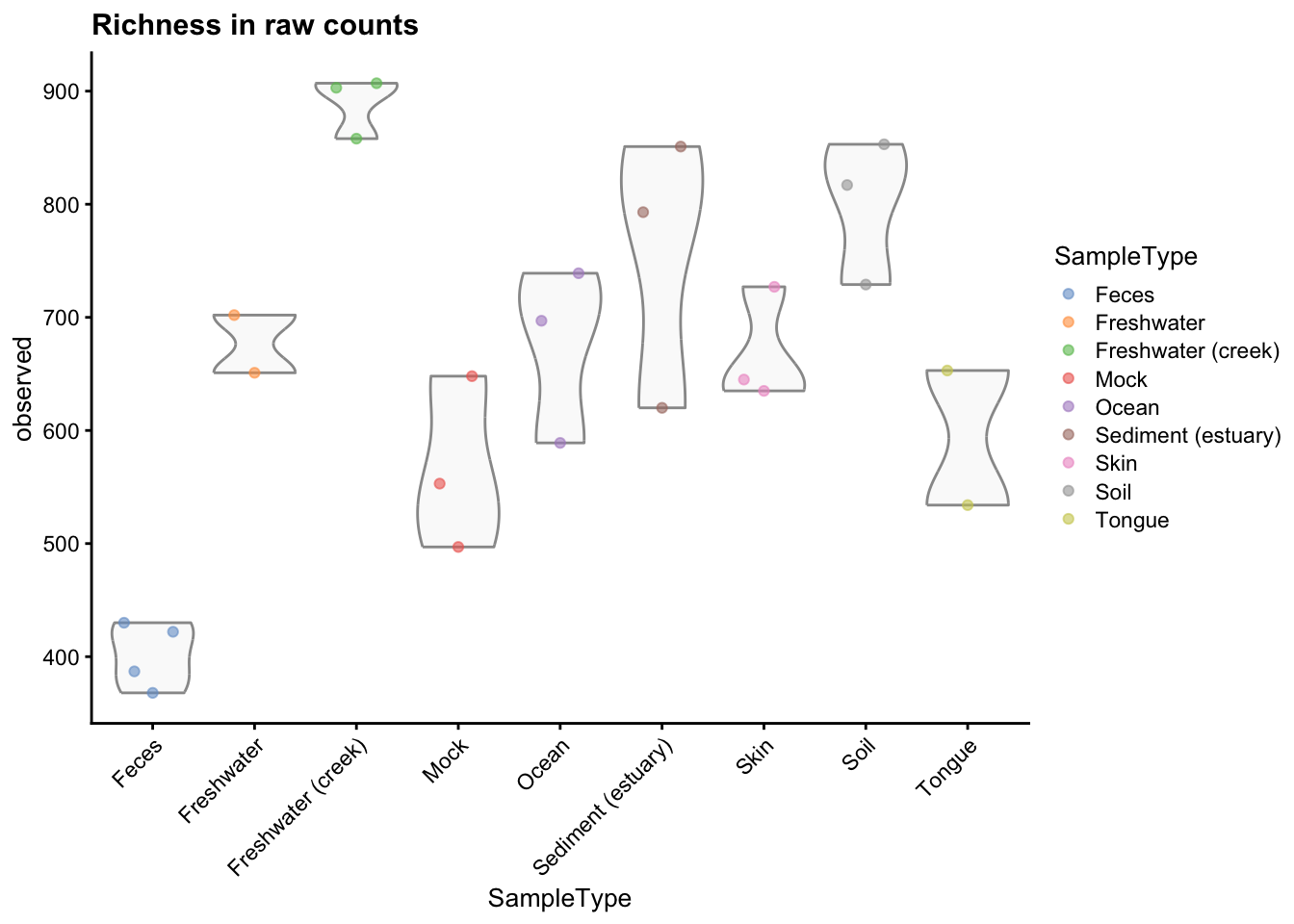
Figure 2.2: Richness estimates grouped and coloured by sample type.
And we can also describe the relationship between library size and richness. When the sequencing depth is enough to describe the samples we do not expect to see any particular pattern in this kind of graphical representation.
plotColData(object = gp_genus,
x = "sum", y = "observed",
colour_by = "SampleType") +
geom_smooth() +
labs(x = "Library Size", y = "Richness",
title = "Richness vs Library Size")## `geom_smooth()` using method = 'loess' and
## formula = 'y ~ x'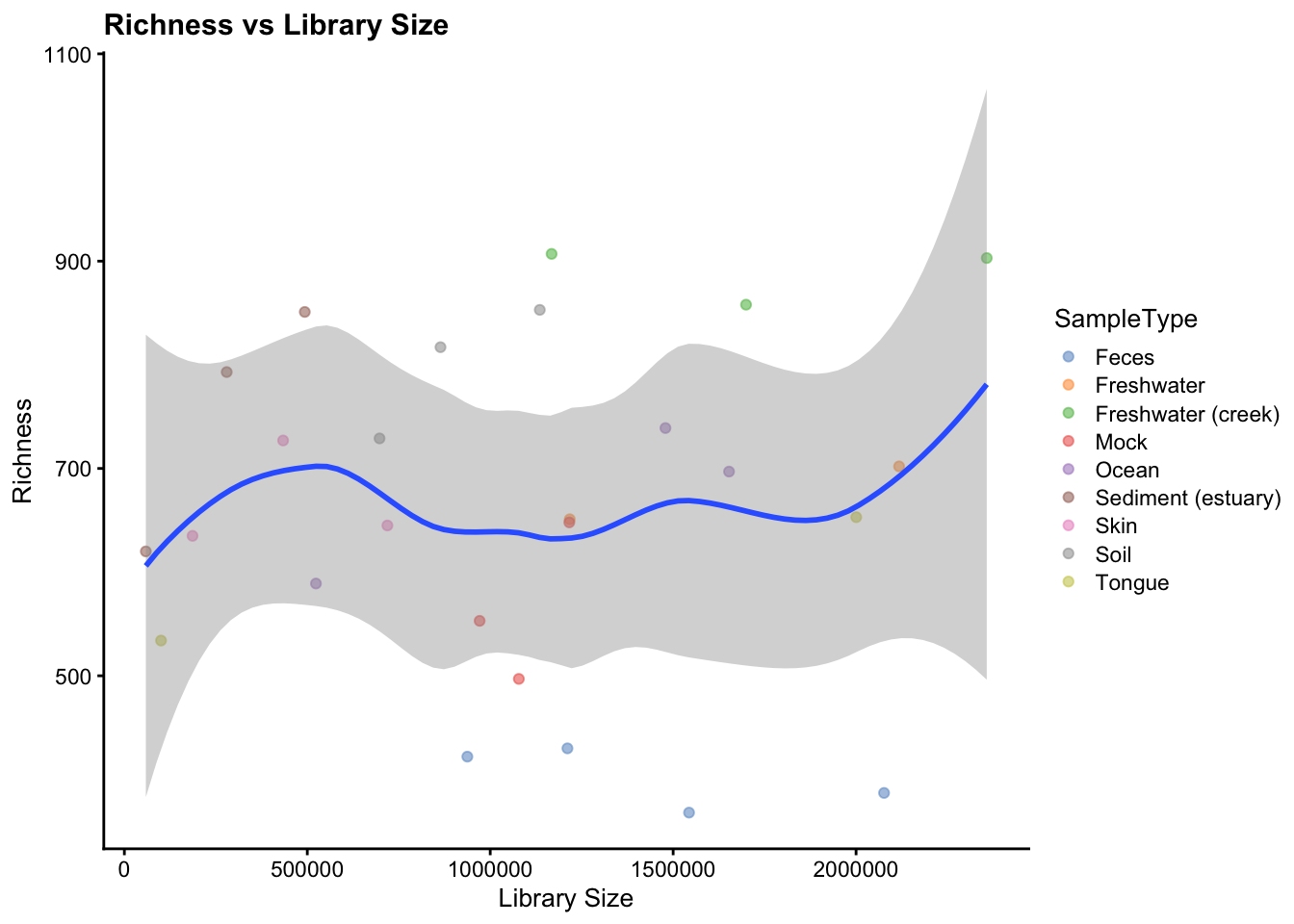
Figure 2.3: Relationship between richness and library size coloured by sample type.
But all of the above plots are generated from raw data. Here we want to inspect the effect of rarefaction. For this reason we use the subsampleCounts function of the package mia to obtain subsampled counts. We repeat this process 3 times with 3 different seeds (1, 2, and 3). We store the results in three differentially named assays of the TreeSummarizedExperiment object: rare1, rare2, and rare3.
gp_genus <- mia::subsampleCounts(gp_genus,
assay.type = "counts",
min_size = min(gp_genus$sum),
seed = 1, replace = FALSE,
verbose = FALSE,
name = "rare1")
gp_genus <- mia::subsampleCounts(gp_genus,
assay.type = "counts",
min_size = min(gp_genus$sum),
seed = 2, replace = FALSE,
verbose = FALSE,
name = "rare2")
gp_genus <- mia::subsampleCounts(gp_genus,
assay.type = "counts",
min_size = min(gp_genus$sum),
seed = 3, replace = FALSE,
verbose = FALSE,
name = "rare3")We compute richness for each subsampled count matrix:
gp_genus <- estimateRichness(gp_genus, assay.type = "rare1", name = "rich1", index = "observed")
gp_genus <- estimateRichness(gp_genus, assay.type = "rare2", name = "rich2", index = "observed")
gp_genus <- estimateRichness(gp_genus, assay.type = "rare3", name = "rich3", index = "observed")And plot the results:
p_r1 <- plotColData(object = gp_genus,
y = "rich1", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Subsampled counts - seed = 1",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
p_r2 <- plotColData(object = gp_genus,
y = "rich2", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Subsampled counts - seed = 2",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
p_r3 <- plotColData(object = gp_genus,
y = "rich3", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Subsampled counts - seed = 3",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
library(patchwork)
(plot_spacer() + p_r + plot_spacer()) / (p_r1 + p_r2 + p_r3) +
plot_layout(guides = "collect") + plot_annotation(tag_levels = "a")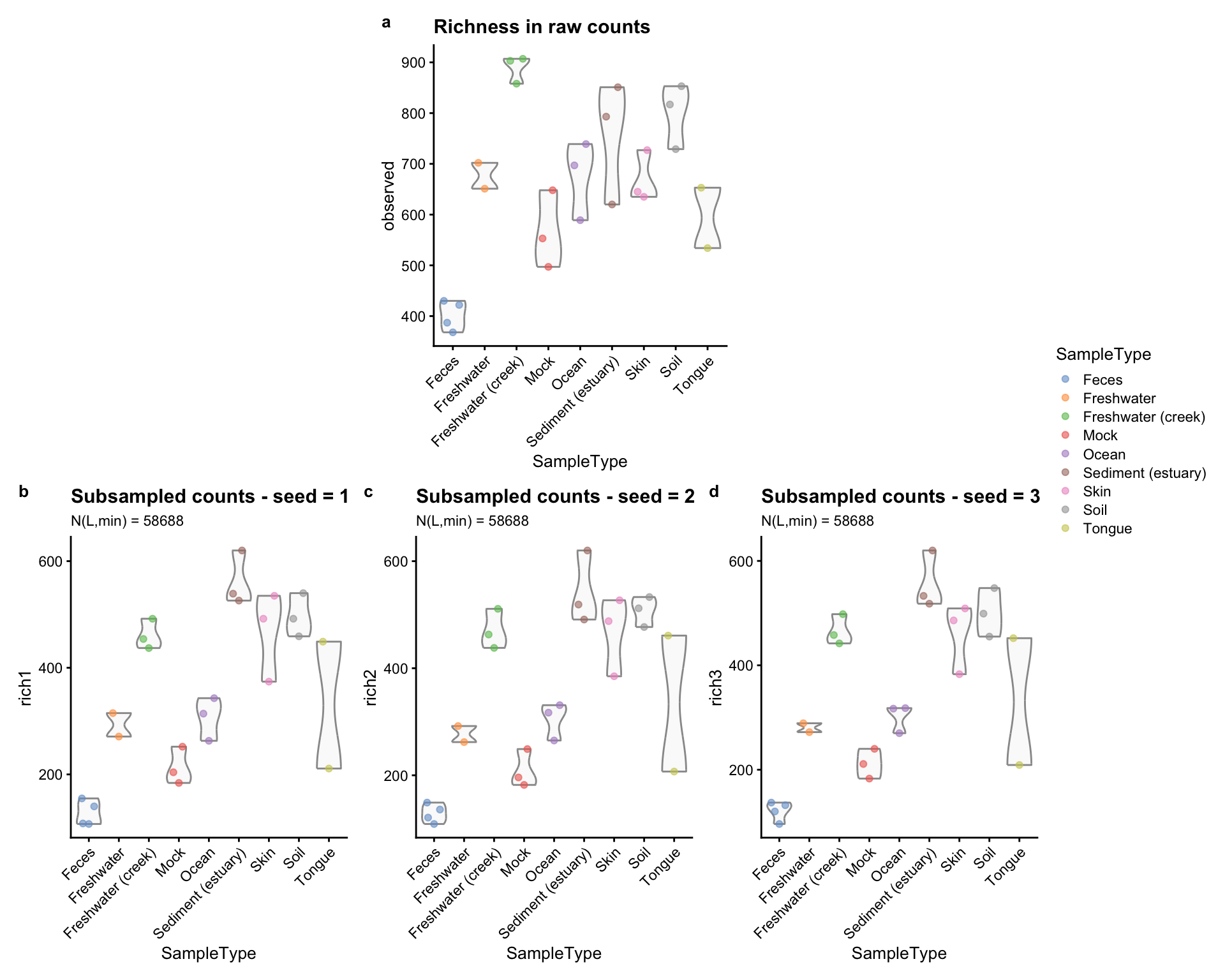
Figure 2.4: Richness estimates grouped and coloured by sample type. a) Raw data. b) subsampled counts, seed = 1. c) subsampled counts, seed = 2. d) subsampled counts, seed = 3.
The main difference in these panels are the range of the y-axis: it reaches higher values in a rather than b, c, and d. Moreover, while the richest samples belonged to Freshwater (creek) when the richness was evaluated using raw counts (panel a), Sediment (estuary) samples became the richest when the subsampled counts were used instead (b, c, and d panels). This is probably due to the differential presence of rare taxa between the two environments. Minor differences are observable when comparing b, c, and d panels, but still, they are present.
Practically, rarefaction consists in averaging the index measures of panels b, c, and d to obtain a single value for each sample.
gp_genus$rich_rarefaction <- rowMeans(
as.data.frame(colData(gp_genus)[,
c("rich1", "rich2", "rich3")]))
p_rarefaction <- plotColData(object = gp_genus,
y = "rich_rarefaction", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Rarefaction Richness",
subtitle = paste("Subsamplings = 3, N(L,min) =",
min(gp_genus$sum)))
p_rarefaction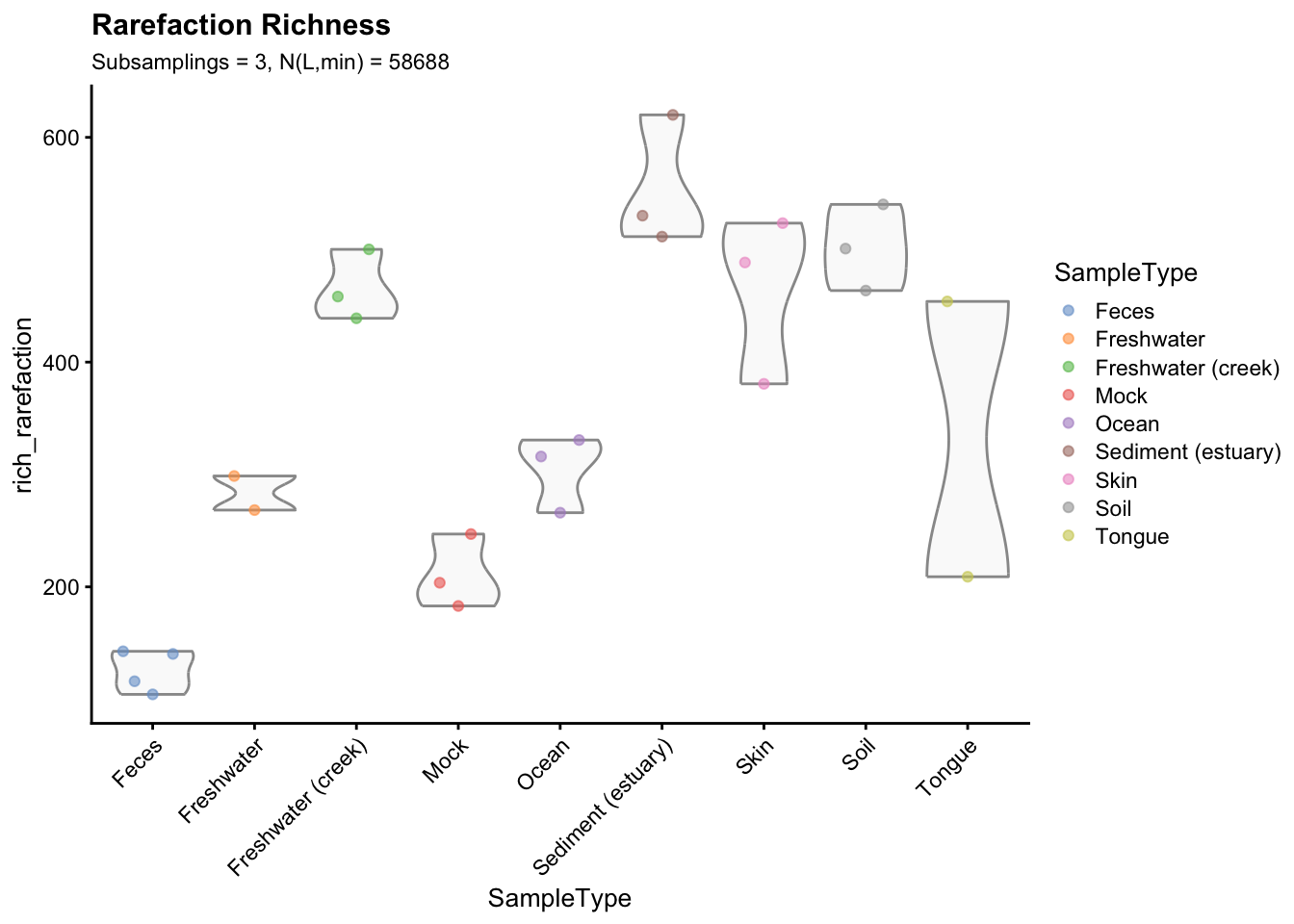
Figure 2.5: Richness estimates grouped and coloured by sample type. Richness indexes were averaged across the 3 subsamplings to obtain a single value for each sample.
To automatise the entire procedure the following function can be used:
library(BiocParallel)
rarefaction_richness <- function(
tse, # The object to use
assay.type = "counts", # Assay to use
subsamplings = 100, # Number of subsamplings
minLibSize = NULL, # The desired library size
seed = 123, # The seed
BPPARAM = SerialParam()){ # Parallelisation
counts <- assay(tse, "counts")
if(is.null(minLibSize))
minLibSize <- min(colSums(counts))
richness_list <- bplapply(
X = 1:subsamplings,
BPPARAM = BPPARAM,
BPOPTIONS = bpoptions(RNGseed = seed),
FUN = function(sub){
# Subsampling step
tse <- mia::subsampleCounts(tse,
assay.type = assay.type,
min_size = minLibSize,
# seed = runif(1, 0, .Machine$integer.max),
replace = FALSE,
verbose = FALSE,
name = "sub")
# Estimation step
richness <- mia::estimateRichness(tse,
assay.type = "sub",
index = "observed",
name = "richness")$richness
return(richness)
})
# Return the averaged values
colMeans(plyr::ldply(richness_list))
}We run it in parallel (Linux or MacOS, Windows users should use BPPARAM = SerialParam()) and we directly store the computed values inside the TreeSummarizedExperiment object.
library("TreeSummarizedExperiment")
gp_genus$rarefaction_richness <- rarefaction_richness(
tse = gp_genus,
assay.type = "counts",
subsamplings = 100,
BPPARAM = MulticoreParam(4),
minLibSize = NULL,
seed = 123)Finally, the graphical representation of rarefied richness (seed = 123, number of subsamplings = 100, \(N_{L,min} = 58688\)) is reported below.
p_rarefaction100 <- plotColData(object = gp_genus,
y = "rarefaction_richness", x = "SampleType",
colour_by = "SampleType") +
theme(axis.text.x = element_text(angle = 45, hjust=1)) +
labs(title = "Rarefaction Richness",
subtitle = paste("Seed = 123, Subsamplings = 100, N(L,min) =",
min(gp_genus$sum)))
p_rarefaction100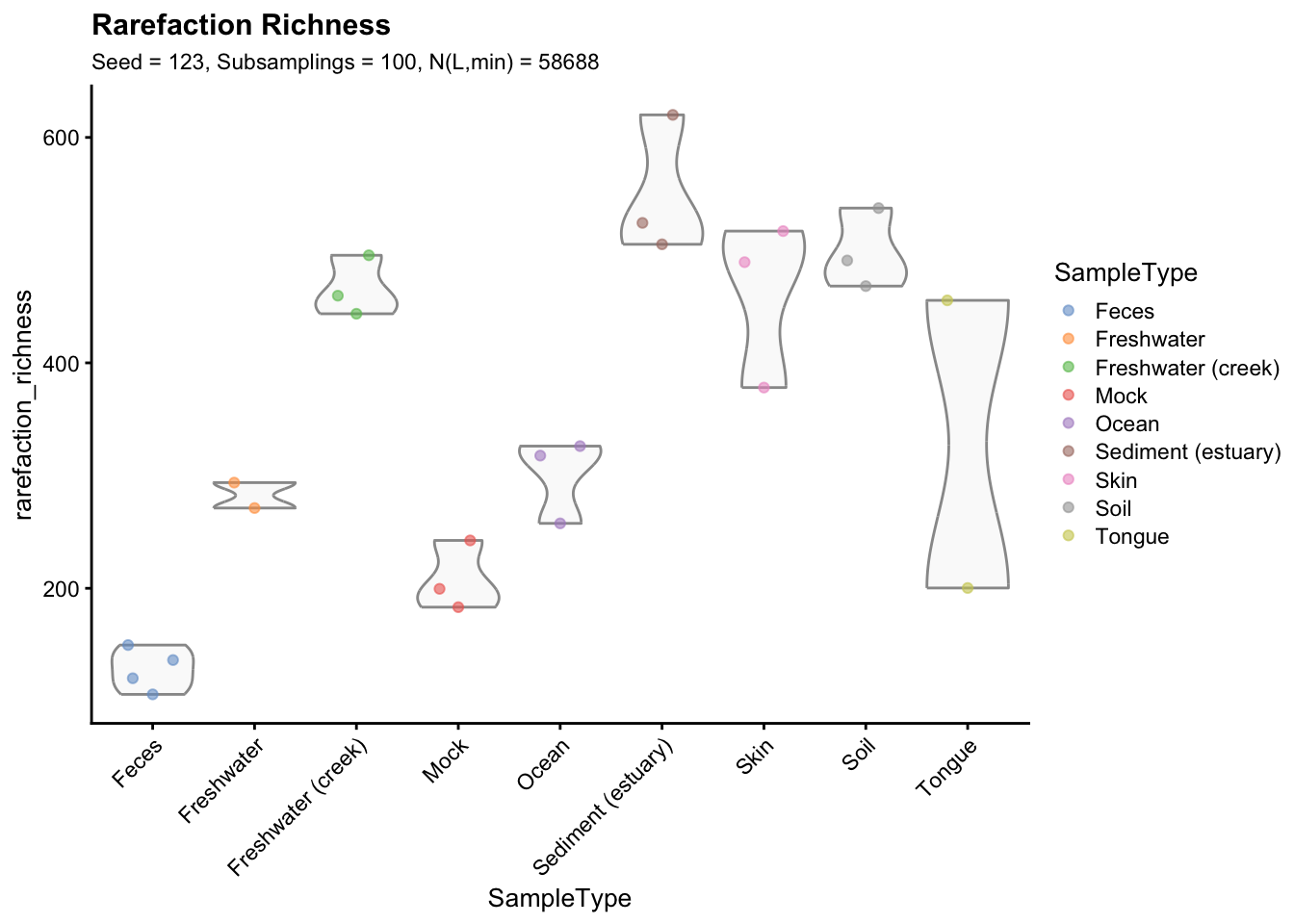
Figure 2.6: Richness estimates grouped and coloured by sample type. Richness indexes were averaged across 100 subsamplings (rarefaction library size = 58688) to obtain a single value for each sample (seed = 123).
2.4.2 Rarefaction curves
One of the most informative application that involves rarefaction and microbial richness is the creation of rarefaction curves:
# devtools::install_github("gauravsk/ranacapa")
library(ranacapa)
set.seed(123)
rarefaction_curves <- ggrare(
mia::makePhyloseqFromTreeSE(gp_genus),
step = 10000, plot = FALSE, parallel = TRUE)library(ggplot2)
# https://rdrr.io/github/LTLA/scater/src/R/plot_colours.R
total_colors <- scater:::.get_palette("tableau10medium")[1:9]
names(total_colors) <- levels(gp_genus$SampleType)
print(rarefaction_curves) +
geom_line(aes(color = SampleType)) +
facet_wrap(~ SampleType, nrow = 3, ncol = 3) +
scale_color_manual(values = total_colors) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(title = "Rarefaction curves",
subtitle = "Faceted by sample type")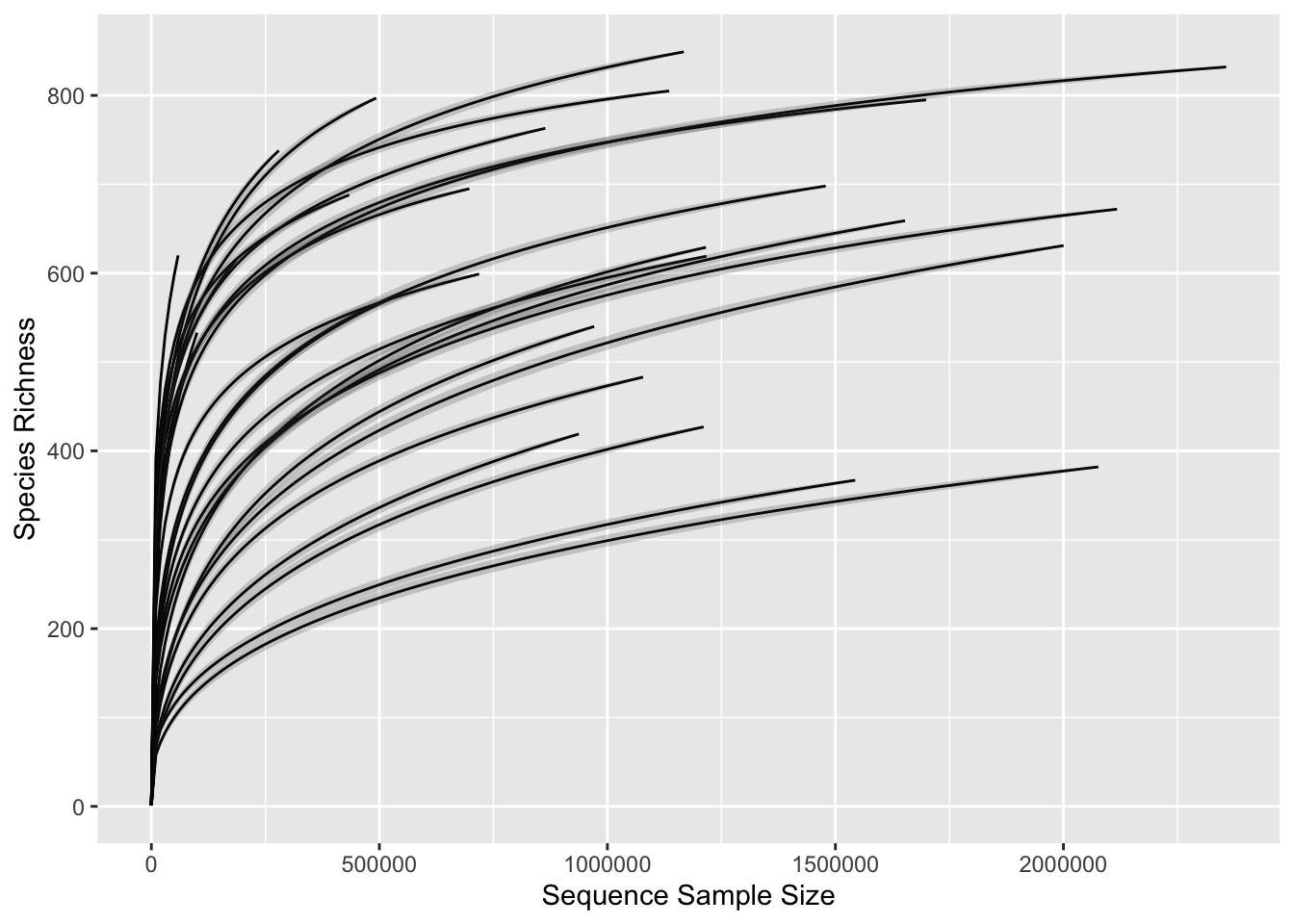
Figure 2.7: Rarefaction curves.
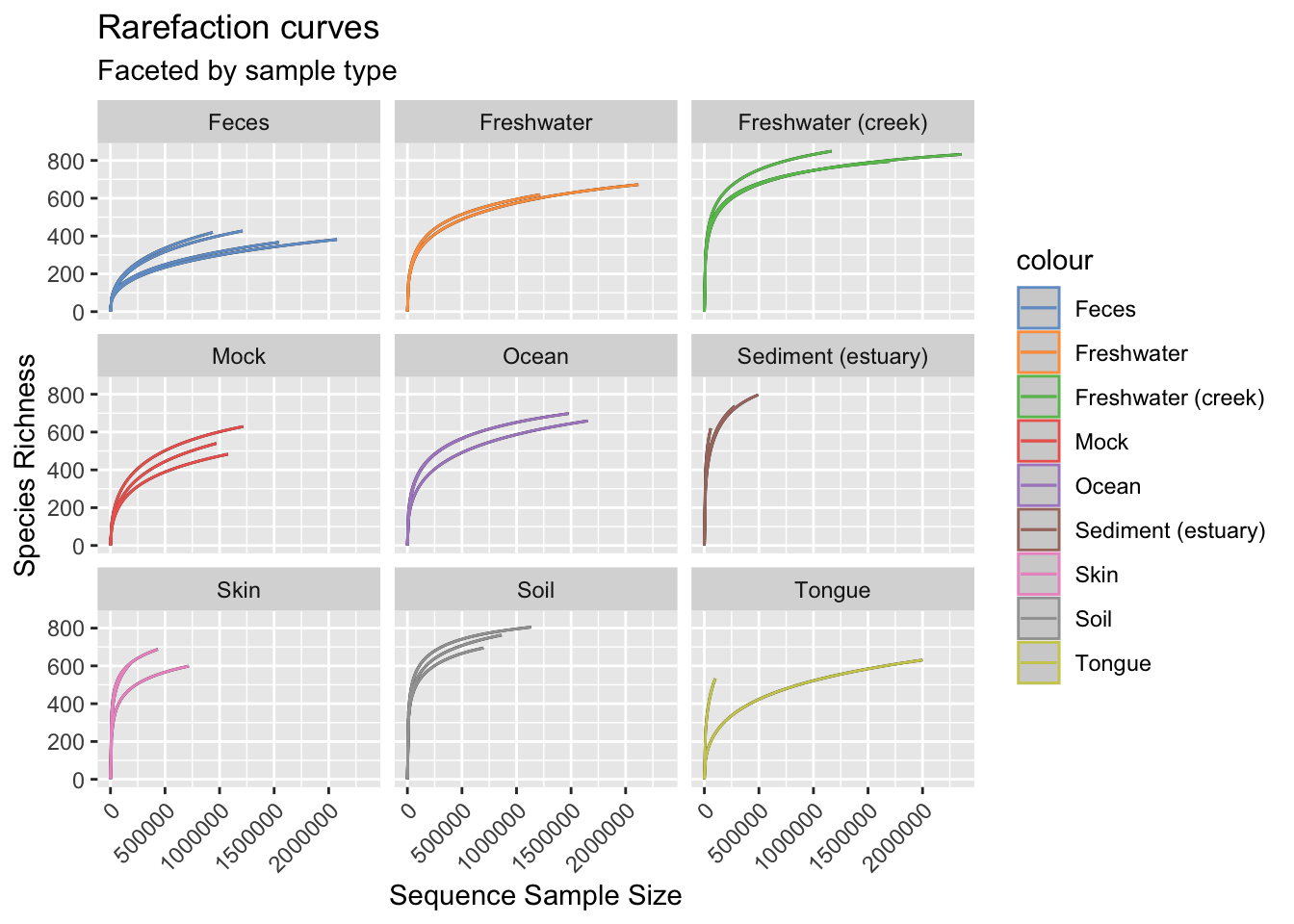
Figure 2.8: Rarefaction curves.
In this case the process of rarefaction consists in repeatedly subsampling the sequencing data at different depths, typically starting from the smallest number of reads in any sample to the largest. At each subsampling depth, the number of observed taxa is calculated (i.e., richness), and these values are plotted against the corresponding sequencing depth. The resulting curve shows how the diversity of the microbial community changes as the sequencing depth increases.
The importance of rarefaction curves lies in several key aspects:
Assessing data completeness. If the curves reach a plateau, it suggests that the sequencing depth is sufficient to capture most of the diversity in the sample. On the other hand, if the curves do not plateau (e.g., one tongue sample, and all Sediment (estuary) samples), it indicates that more sequencing effort is needed to adequately characterize the microbial diversity.
Fair comparison. Rarefaction curves enable fair comparisons of diversity across different samples with varying sequencing depths. By subsampling all samples to the same sequencing depth, the curves provide a standardized view of the community’s diversity, facilitating reliable comparisons.
Assessing quality. Rarefaction curves can help identify samples with low sequencing depth that might require additional attention or potentially be excluded from further analysis due to insufficient data.
Study design optimization. Researchers can use rarefaction curves during the study design phase to estimate the required sequencing depth for reliable and comprehensive community profiling. This information can help optimize cost-effectiveness and resource allocation.
2.4.3 Bray-Curtis dissimilarity index
Similarly to what we did for microbial richness, we can also study the effect of rarefaction on beta-diversity. In particular, we will focus on the Bray-Curtis dissimilarity index, a widely used metric to quantify the dissimilarity or similarity between two ecological samples, typically used in the context of community ecology and biodiversity studies.
Here we start by computing the Bray-Curtis indexes on raw counts, relative abundances (Total Sum Scaling normalization), and some subsamplings (the same used earlier for richness). Then use Multi Dimensional Scaling unsupervised ordination method is used to visualize the results.
gp_genus <- transformCounts(gp_genus,
assay.type = "counts", method = "relabundance")
gp_genus <- runMDS(gp_genus,
FUN = vegan::vegdist,
method = "bray",
name = "PCoA_BC_raw",
assay.type = "counts")
gp_genus <- runMDS(gp_genus,
FUN = vegan::vegdist,
method = "bray",
name = "PCoA_BC_TSS",
assay.type = "relabundance")
gp_genus <- runMDS(gp_genus,
FUN = vegan::vegdist,
method = "bray",
name = "PCoA_BC1",
assay.type = "rare1")
gp_genus <- runMDS(gp_genus,
FUN = vegan::vegdist,
method = "bray",
name = "PCoA_BC2",
assay.type = "rare2")
gp_genus <- runMDS(gp_genus,
FUN = vegan::vegdist,
method = "bray",
name = "PCoA_BC3",
assay.type = "rare3")
p_bc_raw <- plotReducedDim(gp_genus, "PCoA_BC_raw",
colour_by = "SampleType") +
labs(title = "Raw counts, Bray-Curtis, PCoA")
p_bc_tss <- plotReducedDim(gp_genus, "PCoA_BC_TSS",
colour_by = "SampleType") +
labs(title = "TSS, Bray-Curtis, PCoA")
p_bc_r1 <- plotReducedDim(gp_genus, "PCoA_BC1",
colour_by = "SampleType") +
labs(title = "Subsampled seed = 1, Bray-Curtis, PCoA",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
p_bc_r2 <- plotReducedDim(gp_genus, "PCoA_BC2",
colour_by = "SampleType") +
labs(title = "Subsampled seed = 2, Bray-Curtis, PCoA",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
p_bc_r3 <- plotReducedDim(gp_genus, "PCoA_BC3",
colour_by = "SampleType") +
scale_y_reverse() +
labs(title = "Subsampled seed = 3, Bray-Curtis, PCoA",
subtitle = paste("N(L,min) =", min(gp_genus$sum)))
(p_bc_raw + p_bc_tss + plot_spacer()) /
(p_bc_r1 + p_bc_r2 + p_bc_r3) +
plot_layout(guides = "collect") +
plot_annotation(tag_levels = "a")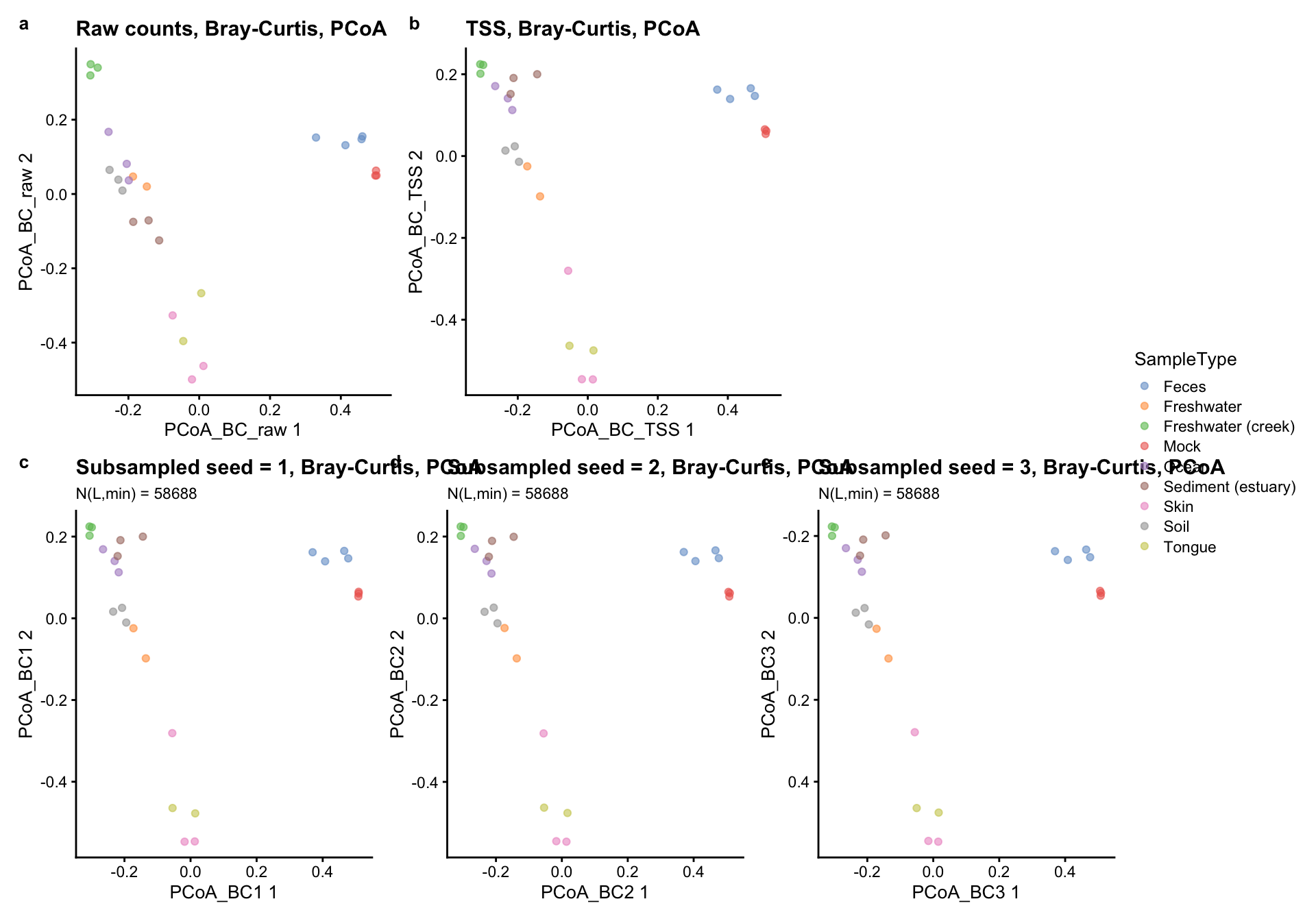
The main difference in these panels is only between a and all the others which are quite similar. That’s because in this case the total sum scaling normalization (panel b) behave similarly to using subsampling at the fixed library size of \(N_{L,min}=58688\) (panels c, d, and e).
The Bray-Curtis dissimilarity between two samples is computed following the formula:
\[ BC_{jk} = \frac{\sum_i|x_{ij}-x_{ik}|}{\sum_ix_{ij}+x_{ik}} \]
where \(i\) is the number of taxa and \(j,k\) are the sample indexes. By construction, when a sample has 10x more counts than the other, the numerator could be inflated just for this reason and not because of a real difference between the samples.
Practically, rarefaction consists in averaging the index measures of panels b, c, and d to obtain a single value for each sample.
# Recompute the distances
bc1 <- vegan::vegdist(x = t(assay(gp_genus, "rare1")),
method = "bray")
bc2 <- vegan::vegdist(x = t(assay(gp_genus, "rare2")),
method = "bray")
bc3 <- vegan::vegdist(x = t(assay(gp_genus, "rare3")),
method = "bray")
# Average the distances across subsamplings
bc_rarefaction <- (bc1 + bc2 + bc3) / 3
# Add the PCoA coordinates in a new reducedDim slot
# following the instructions in help("runMDS")
reducedDim(gp_genus, "PCoA_BCrarefaction") <- cmdscale(d = bc_rarefaction, k = 2, eig = TRUE)$pointsplotReducedDim(gp_genus, "PCoA_BCrarefaction",
colour_by = "SampleType") +
labs(title = "Rarefaction Bray-Curtis, PCoA",
subtitle = paste("subsamplings = 3, N(L,min) =", min(gp_genus$sum)))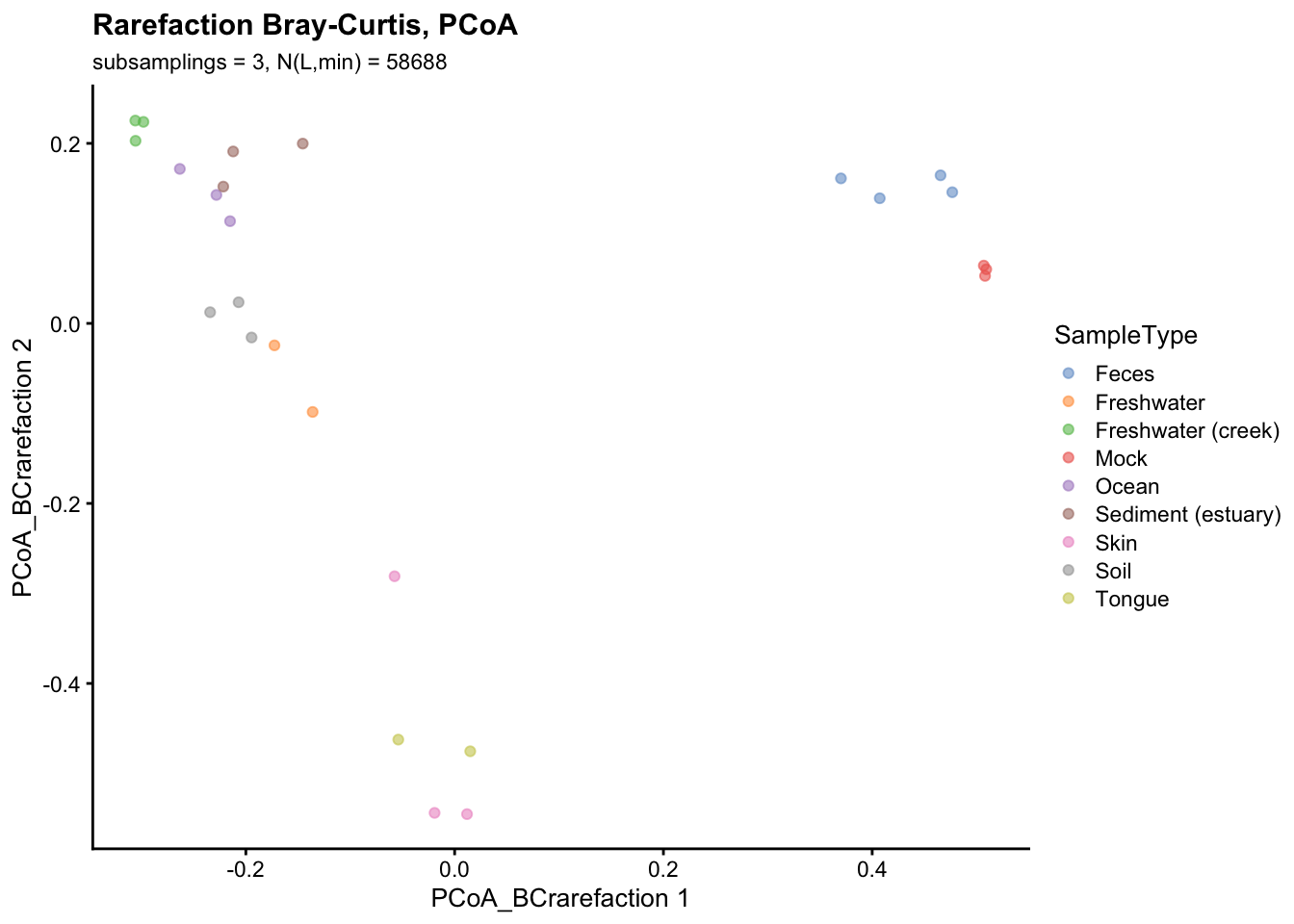
As we did for the richness index, the process can be automatised:
rarefaction_BC_PCoA <- function(
tse, # The object to use
assay.type = "counts", # Assay to use
subsamplings = 10, # Number of subsamplings
minLibSize = NULL, # The desired library size
seed = 123, # The seed
BPPARAM = SerialParam()){ # Parallelisation
counts <- assay(tse, "counts")
if(is.null(minLibSize))
minLibSize <- min(colSums(counts))
dist_list <- bplapply(
X = 1:subsamplings,
BPPARAM = BPPARAM,
BPOPTIONS = bpoptions(RNGseed = seed),
FUN = function(sub){
# Subsampling step
tse <- mia::subsampleCounts(tse,
assay.type = assay.type,
min_size = minLibSize,
# seed = runif(1, 0, .Machine$integer.max),
replace = FALSE,
verbose = FALSE,
name = "sub")
# BC calculation step
BC_dist <- vegan::vegdist(
x = t(assay(tse, "sub")),
method = "bray")
return(BC_dist)
})
# Compute the mean BC distance
avg_BC_dist <- Reduce(f = '+', dist_list) / subsamplings
# Compute the PCoA coordinates and return them
cmdscale(d = avg_BC_dist, k = 2, eig = TRUE)$points
}We run it in parallel (Linux or MacOS, Windows users should use BPPARAM = SerialParam()) and we directly store the computed values inside the TreeSummarizedExperiment object in the reducedDim slot.
reducedDim(gp_genus, "PCoA_BCrarefaction100") <-
rarefaction_BC_PCoA(
tse = gp_genus,
assay.type = "counts",
subsamplings = 100,
minLibSize = NULL,
seed = 321,
BPPARAM = MulticoreParam(4))Finally, the graphical representation of rarefied beta-diversity using Bray-Curtis dissimilarity index and PCoA ordination (seed = 123, number of subsamplings = 100, \(N_{L,min} = 58688\)) is reported below.
plotReducedDim(gp_genus, "PCoA_BCrarefaction100",
colour_by = "SampleType") +
labs(title = "Rarefaction Bray-Curtis, PCoA",
subtitle = paste("subsamplings = 100, seed = 321, N(L,min) =", min(gp_genus$sum))) +
scale_y_reverse()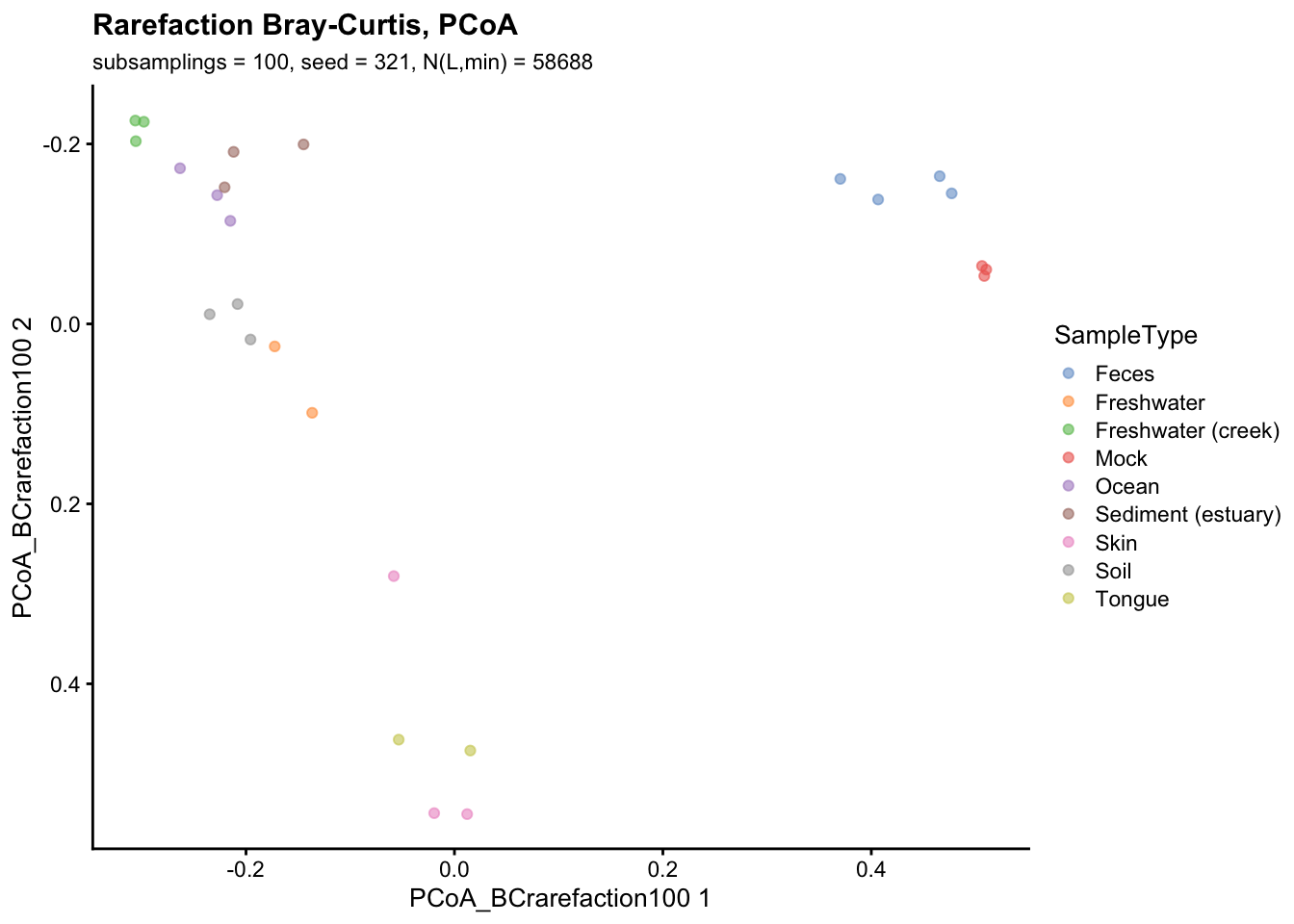
In this scenario rarefaction and Total Sum Scaling (TSS) normalization produced similar results. This is probably due to the fact that all samples are already at a sufficient depth to capture most of the microbial diversity.
But this can also occur when:
the differences in sequencing depth between samples are relatively small;
the samples have similar distributions of taxa and similar alpha diversity measures, so the impact of rarefaction or TSS normalization is minimal.
However, in cases where there are substantial differences in sequencing depth or if the samples have highly variable distributions of taxa, rarefaction and TSS normalization might lead to different results. In such situations, researchers should carefully consider which normalization approach is more appropriate for their specific dataset and research objectives.
2.5 Exercise: loading real datasets
To test R packages it’s convenient to rely on high-quality, polished datasets such as GlobalPatterns. In the real world, though, we need to load datasets produced by different packages (often in different formats), and to produce the appropriate data structures (e.g. a Phyloseq object) starting from our raw files.
In addition to a lot of data wrangling and format conversions, real data is noisy, contains errors or inconsistencies and we need to be able to perform some checks and eyeballing or we will miss the gorilla.
We have some files produced analysing the famous “MiSeq SOP” dataset from the laboratory of Pat Schloss himself (Schloss et al., 2012), based on a longitudinal study of gut microbiome stabilization in mice post-weaning.
The files analised here are available in the Github repository of this chapter:
- dada2_stats.tsv: a summary of the performance of DADA2 denoising
- feature-table.tsv: raw counts per feature (ASVs) per sample
- metadata.tsv: properties of each sample
- rep-seqs.tree: phylogenetic tree of the ASVs
- taxonomy.txt: table with the taxonomic classification of each ASV
For the purpose of this chapter, we will not delve into the specific details on the reads to counts procedure, which will be discussed elsewhere soon.
2.5.1 Read the raw data
The pipeline used to identify the Amplicon Sequence Variants (ASV) is based on DADA2, and produced a table with some statistics on the procedure, called dada2_stats.tsv.
Ignoring similar tables can produce flawed analyses: sometimes only a very small fractions of reads passes all the filtering steps, so from a sequencing depth of 50.000 you can generate a counts table where the total is reduced to 1000. Such reduction must be addressed before doing any analysis.
# Read dada2 stats file
quality_stats <- read.delim("./data/01_rarefaction/dada2_stats.tsv", header = TRUE, sep = "\t")
# Add rownames
rownames(quality_stats) <- gsub(pattern = "_R1.fastq.gz", replacement = "", x = quality_stats[,"X"])
# Remove the first column
quality_stats <- quality_stats[, -1]
head(quality_stats)## input filtered denoised merged non.chimeric
## F3D0 7793 6302 6302 5636 5627
## F3D1 5869 4616 4616 4314 4304
## F3D141 5958 4897 4897 4221 4087
## F3D142 3183 2631 2631 2157 2113
## F3D143 3178 2624 2624 2193 2172
## F3D144 4827 3751 3751 3036 2882Some kind of metadata file is usually part of the analysis, we have a minimal metadata file called metadata.tsv:
# Read metadata
metadata <- read.delim("./data/01_rarefaction/metadata.tsv", header = TRUE, sep = "\t", row.names = 1)
head(metadata)## Week Files
## F3D0 Early F3D0_S188_L001_R1_001.fastq.gz,F3D0_S188_L001_R2_001.fastq.gz
## F3D1 Early F3D1_S189_L001_R1_001.fastq.gz,F3D1_S189_L001_R2_001.fastq.gz
## F3D141 Late F3D141_S207_L001_R1_001.fastq.gz,F3D141_S207_L001_R2_001.fastq.gz
## F3D142 Late F3D142_S208_L001_R1_001.fastq.gz,F3D142_S208_L001_R2_001.fastq.gz
## F3D143 Late F3D143_S209_L001_R1_001.fastq.gz,F3D143_S209_L001_R2_001.fastq.gz
## F3D144 Late F3D144_S210_L001_R1_001.fastq.gz,F3D144_S210_L001_R2_001.fastq.gzAn algorithm has been used to assign the taxonomy to the identified representative sequences. The taxonomy table is called taxonomy.tsv.
# Read taxonomy
taxonomy <- read.delim("./data/01_rarefaction/taxonomy.txt", header = TRUE, sep = " ")
rownames(taxonomy) <- paste0("ASV", seq_len(nrow(taxonomy)))
head(taxonomy)## Kingdom Phylum Class Order Family
## ASV1 Bacteria <NA> <NA> <NA> <NA>
## ASV2 <NA> <NA> <NA> <NA> <NA>
## ASV3 Bacteria Bacteroidota Bacteroidia Bacteroidales unclassified_Bacteroidales
## ASV4 <NA> <NA> <NA> <NA> <NA>
## ASV5 Bacteria Bacteroidota Bacteroidia Bacteroidales Bacteroidaceae
## ASV6 <NA> <NA> <NA> <NA> <NA>
## Genus
## ASV1 <NA>
## ASV2 <NA>
## ASV3 <NA>
## ASV4 <NA>
## ASV5 Bacteroides
## ASV6 <NA>Finally, the central piece of information is the feature table (historically called “OTU Table”):
# Read feature table
feature_table <- read.delim("./data/01_rarefaction/feature-table.tsv", header = TRUE, sep = "\t", row.names = 1)
head(feature_table)## F3D0 F3D1 F3D141 F3D142 F3D143 F3D144 F3D145 F3D146 F3D147 F3D148 F3D149
## ASV1 556 377 422 279 216 387 609 311 1407 807 825
## ASV2 322 334 344 282 166 254 466 223 1127 676 735
## ASV3 416 210 319 149 185 281 472 233 860 548 676
## ASV4 402 64 455 155 221 325 539 376 1017 791 829
## ASV5 148 127 175 162 120 93 277 159 424 405 382
## ASV6 439 40 313 178 226 333 447 260 1091 818 599
## F3D150 F3D2 F3D3 F3D5 F3D6 F3D7 F3D8 F3D9 Mock
## ASV1 298 3292 938 303 961 602 263 496 0
## ASV2 216 1486 560 256 643 488 333 403 0
## ASV3 381 1111 437 264 542 397 331 455 0
## ASV4 434 441 192 150 373 290 128 191 0
## ASV5 158 311 375 143 436 419 509 552 0
## ASV6 200 111 25 19 17 9 0 0 0Sometimes it’s useful to know how related are the representative sequences, and this is usually done producing a multiple alignment from which a phylogenetic tree can be constructed.
An example of use of phylogenetic information is the computation of UniFrac distance.
# Read tree file
library(ape)
tree <- ape::read.tree(file = "./data/01_rarefaction/rep-seqs.tree")
str(tree)## List of 5
## $ edge : int [1:443, 1:2] 224 225 226 227 228 229 230 230 229 231 ...
## $ edge.length: num [1:443] 0.00906 0.00835 0.01424 0.00472 0.01043 ...
## $ Nnode : int 221
## $ node.label : chr [1:221] "" "0.929" "0.764" "0.846" ...
## $ tip.label : chr [1:223] "ASV147" "ASV202" "ASV66" "ASV61" ...
## - attr(*, "class")= chr "phylo"
## - attr(*, "order")= chr "cladewise"2.5.2 Assembling the TreeSummarizedExperiment object
tse <- TreeSummarizedExperiment(
assays = list("counts" = feature_table),
rowData = taxonomy,
colData = merge(metadata, quality_stats, by = 0),
rowTree = tree)
tse## class: TreeSummarizedExperiment
## dim: 223 20
## metadata(0):
## assays(1): counts
## rownames(223): ASV1 ASV2 ... ASV222 ASV223
## rowData names(6): Kingdom Phylum ... Family Genus
## colnames(20): F3D0 F3D1 ... F3D9 Mock
## colData names(8): Row.names Week ... merged non.chimeric
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):
## rowLinks: a LinkDataFrame (223 rows)
## rowTree: 1 phylo tree(s) (223 leaves)
## colLinks: NULL
## colTree: NULL2.5.3 Compute quality metrics
For both the colData (i.e., sample metadata) and rowData (i.e., taxa metadata) slots of the experiment object we calculate some simple metrics. For the samples, the library size (sum) and the number of detected features (detected) are computed:
tse <- scater::addPerFeatureQC(tse)
tse <- scater::addPerCellQC(tse)
head(colData(tse)[, c("sum", "detected")])## DataFrame with 6 rows and 2 columns
## sum detected
## <numeric> <numeric>
## F3D0 5627 103
## F3D1 4304 94
## F3D141 4087 71
## F3D142 2113 49
## F3D143 2172 57
## F3D144 2882 45For the taxa, their average count value (mean) and the percentage of samples with that feature (detected) are reported:
## DataFrame with 6 rows and 2 columns
## mean detected
## <numeric> <numeric>
## ASV1 667.45 95
## ASV2 465.70 95
## ASV3 413.35 95
## ASV4 368.65 95
## ASV5 268.75 95
## ASV6 256.25 852.5.4 Filter or not to filter, this is the question
When we loaded the raw data, we may have noticed that many taxa were not classified, even at the kingdom and phylum level. In particular we have 20 ASVs which are not classified at the Kingdom level and 30 (the previous 20 plus other 10) at the Phylum level.
##
## Bacteria <NA>
## 203 20##
## Actinobacteriota Bacteroidota Campilobacterota
## 6 5 1
## Cyanobacteria Deinococcota Firmicutes
## 3 1 166
## Patescibacteria Proteobacteria unclassified_Bacteria
## 2 7 1
## Verrucomicrobiota <NA>
## 1 30Whether or not to remove unclassified features at the phylum level prior to every analysis in microbiome data depends on the specific goals and requirements of the analysis.
The presence of unclassified features may be due to various reasons, such as limited reference databases or low sequencing depth. On one hand, removing these unclassified features could improve data quality and reduce potential noise in your analysis. On the other hand, The decision to remove unclassified features may affect the outcome of the analysis. If the unclassified features are prevalent and significant in your dataset, removing them could lead to a loss of valuable information. Nevertheless, if they are rare or not biologically relevant, removing them might have minimal impact.
## DataFrame with 30 rows and 2 columns
## mean detected
## <numeric> <numeric>
## ASV1 667.45 95
## ASV2 465.70 95
## ASV4 368.65 95
## ASV6 256.25 85
## ASV7 254.85 95
## ... ... ...
## ASV193 0.55 5
## ASV195 0.50 20
## ASV196 0.50 5
## ASV201 0.40 5
## ASV204 0.30 10In our case we can see that some of the unclassified features are in the top abundant positions. These are hardly related to rare features as they are present with high mean abundances in more than 90% of the samples.
Whatever the choice, if you plan to share your data or replicate the analysis in the future, it’s essential to document clearly whether unclassified features were removed and the reasoning behind the decision.
2.5.5 Try again?
A closer inspection of the data raised an issue: multiple features (ASVs) had no taxonomy attached.
We noticed that high abundance and high prevalence features were not immune from the issue. If we can be sure that our taxonomy assignment was flawless, we might conclude we are observing new sequences, and it would be advisable to perform the analysis at the feature level, ignoring taxonomic lables when possible.
An alternative is to check our methods, and redo the taxonomy classification. Try loading taxonomy-dada.txt and you will now see a different story…
Bibliography
Quadram Institute Bioscience, andrea.telatin@quadram.ac.uk↩︎